



















The challenges of medical device software development extend beyond what is required for other types of healthcare software and technology, because the software itself is a medical device in its own right. Therefore, the final product has to adhere to both standards on healthcare software and standards on a hardware medical device.
Being an external development team that helps clients develop their SaMD products, we are sure that working with an outside team of engineers is often the key to the delivery of quality healthcare software products. Throughout our experience, we have learned how to watch for and navigate a slew of potential pitfalls, which is invaluable since we constantly work on new technologies and products for the healthcare industry.
In this article, we will take a look at the challenges facing companies when it comes to creating and developing SaMD products. We will also explore the opportunities for companies that engage an external development team in order to create, update, and maintain their product.
Developing SaMD is a challenging and complicated process, especially in terms of strict regulation requirements that govern this type of software. It requires a tech team that understands what is at stake. Just as with any medical devices and equipment, software as a medical device is actively used in diagnosis and treatment and is therefore subject to strict standards.
There are a number of standards and regulations addressing SaMD development. Some of these standards are in place globally, while others apply to a specific market region. There are several international standards that developers have to take into account in order to develop safe and effective health software products.
The International Medical Device Regulators Forum (IMDRF) and U.S. Food and Drug Administration (FDA) work together to enable SaMD development companies to advance safe technologies and ensure patient safety.
SaMD regulation is not one-size-fits-all. Knowing the regulatory standards and how they apply to a particular product is one of the first steps in the process of successful SaMD development and deployment.
Below are top three international standards that companies and their development teams should consider prior to submitting any documentation for market approval:
Understanding the product’s risk class is crucial for determining the scope and the rigor of the regulatory requirements medical device software has to comply with. The medical device risk classification is assigned based on the intended use, risks associated with the device, and the levels of controls necessary to ensure its effectiveness and safety.
In the US, the FDA offers its own guidance on how to determine the risk class for a SaMD. The FDA categorizes medical devices into three classes:
In the EU, it is the European Union Medical Device Regulation, or EU MDR 2017/745, that covers medical device classification. It classifies the devices as follows:
Basically, the higher the class is, the stricter the requirements are in terms of the device’s safety, performance, and effectiveness.
Methods for risk classification vary depending on the market region and regulatory organizations. The same medical device may be considered a low-risk device in the US and exempted from premarket submissions, but a high risk device in the EU and therefore subject to the review of regulatory bodies. Familiarity with these complex classifications helps optimize delivery to market.
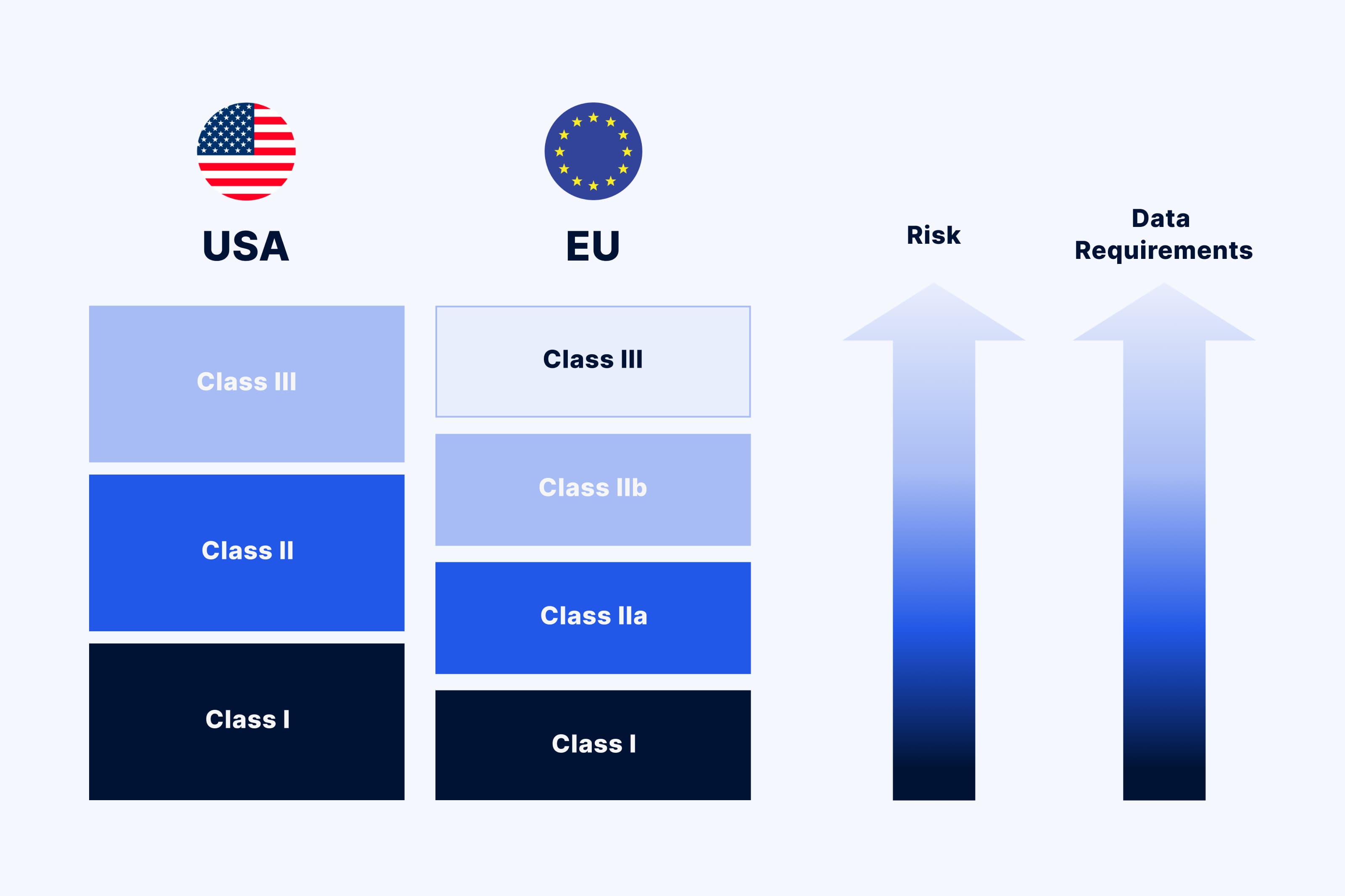
SaMD classification
The processes of verification and validation are crucial to regulatory compliance, patient safety, and high-quality medical device software development. They are an integral part of a quality management system. Adhering to the verification and validation processes from the start helps improve applicability, accelerate time to market, and reduce production costs. Quality management also reduces the chances of errors that might result in expensive and time-consuming rework and redesign.
Verification comes first and ensures that the system requirements have been implemented correctly. This will generally include not only different types of testing (unit testing, integration testing, etc.), but also static and dynamic analysis, code reviews, walkthroughs, and document inspections.
The aim of software validation is to check whether the actual product satisfies the predefined business goals and users’ needs. This is achieved through a wide array of activities such as human factors testing, analysis and inspection methods. Clinical evaluation is also an important part of the validation process. It is used to collect and assess clinical data relating to a medical device to ensure that there is sufficient evidence that the device conforms to the necessary regulatory requirements.
In accordance with the FDA, clinical evaluation of SaMD includes three key components:
Gathering accurate data from these software validation processes will help you strengthen your path to regulatory compliance and reduce potential pitfalls when developing a product.
Regardless of the market region, be it the U.S., the EU, etc., SaMD manufacturers face formal inspections by regulatory agencies. The goal of such inspections is to ensure the QS’s compliance with the certain regulations and thus to protect the market from unsafe products.
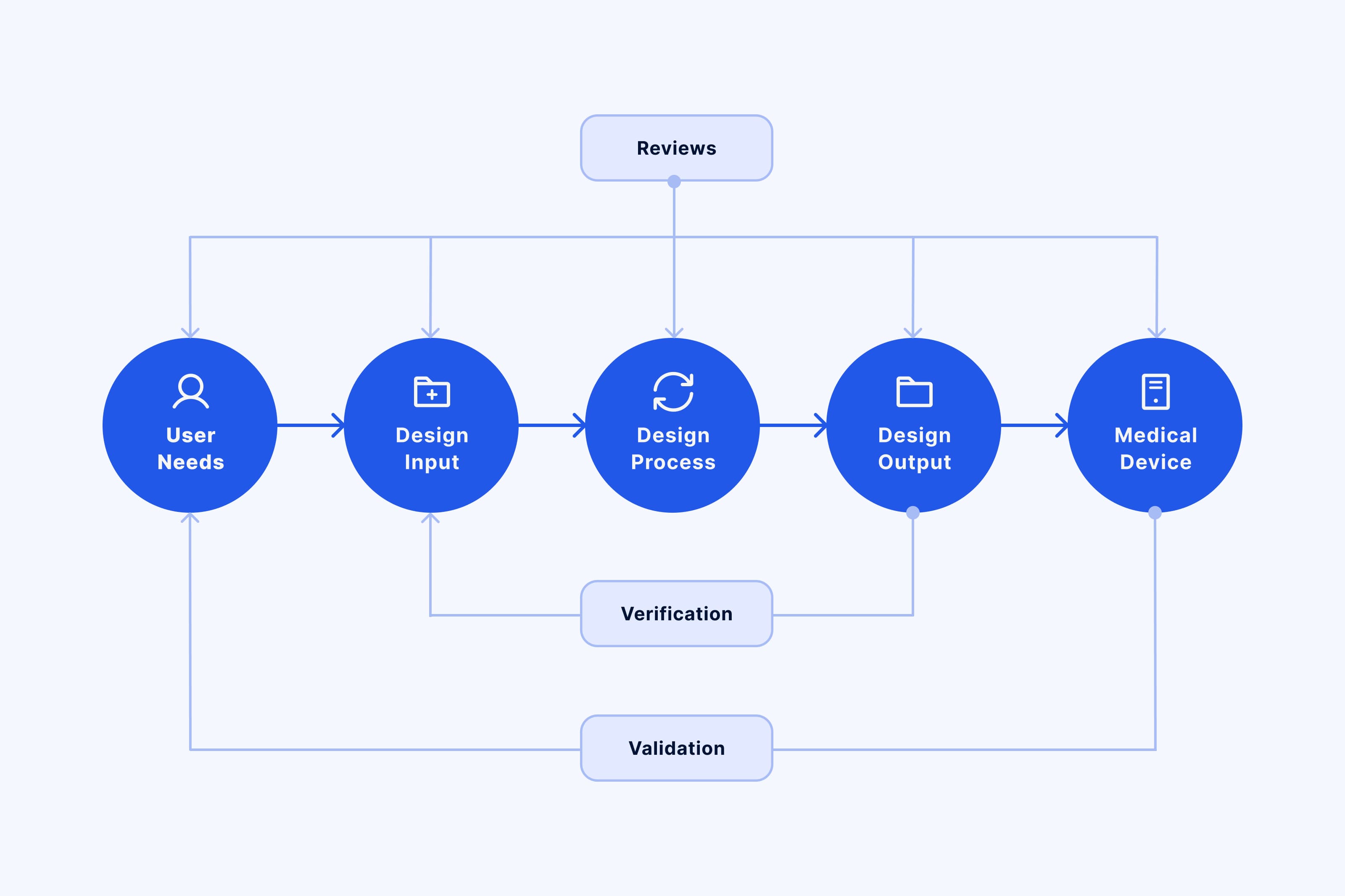
Design controls in SaMD development
There are two major regulations to consider in terms of QMS:
It is important to choose the right quality system as it not only ensures that you keep up with the necessary regulatory requirements but also serves your company’s needs in planning, designing, implementing, and distributing SaMD products.
SaMD developers face all typical challenges associated with development, as well as all additional burdens of medical standards, verification and validation. Besides providing a smooth and seamless SaMD development process, our experience of working with teams in the SaMD space has given us strategies for delivering a safe and effective product. Below are a few strategies we consider fundamental for successful SaMD development.
Because of the heavy burden presented by FDA compliance, there is a common misconception that SaMD developers must stick with the waterfall project methodology, using a linear and sequential framework for development.
However, FDA requirements do not demand any certain methodology, and the FDA itself recognizes that the waterfall approach is not always an option and mainly suits highly stable and structured projects. What is more, it has recognized the agile software development methodology as an acceptable and valid approach for medical device software development. This is perfectly reasonable since functional requirements often change throughout the course of a project. Moreover, the flexibility and possibility of making changes to the system at any development stage are particularly important to keep up with innovative technologies. At EffectiveSoft, we provide a case-by-case approach, considering the requirements and unique characteristics of each project.
Proper data collection and documentation are crucial when dealing with SaMD solutions that have to go through clinical trials and withstand rigid regulations. A QMS is the main tool that development and engineering teams use to ensure adherence to the procedures required throughout the SaMD development life cycle.
Without a robust QMS, you may fail to effectively meet the regulatory requirements for medical devices. This can lead to increased time to market and expensive redevelopment. The QM solutions we provide here at EffectiveSoft encompass quality control, quality assurance, and total quality management.
Building the development process around medical device software requirements is the key to a successful product release. We integrate a quality system that allows managing software documentation, processes, and procedures to meet quality requirements and enhance business workflows.
We recommend working with an external development team that is experienced in software as a medical device development in order to navigate potential pitfalls and successfully bring your product to the market. With extensive experience in software design for medical devices, EffectiveSoft can help you identify the right regulatory strategy to enter the medical software market smoothly and with fewer risks.
*In this article, the terms software as a medical device (SaMD) and medical device software (MDSW) are used interchangeably.
Blog


























Our team would love to hear from you.
Fill out the form, and we’ve got you covered.
What happens next?
San Diego, California
4445 Eastgate Mall, Suite 200
92121, 1-800-288-9659
San Francisco, California
50 California St #1500
94111, 1-800-288-9659
Pittsburgh, Pennsylvania
One Oxford Centre, 500 Grant St Suite 2900
15219, 1-800-288-9659
Durham, North Carolina
RTP Meridian, 2530 Meridian Pkwy Suite 300
27713, 1-800-288-9659
San Jose, Costa Rica
C. 118B, Trejos Montealegre
10203, 1-800-288-9659

